
Самое частое из редких: что нужно знать о СМА
Спинальная мышечная атрофия
Врачи объединяют несколько видов наследственных заболеваний, характеризующихся нарушением движения, в одну группу под названием спинальная мышечная атрофия. В МКБ-10 они идут под кодом G12 с дополнительными указаниями на тип болезни.
По данным исследователей, около 0,01-0,02% детей рождаются с диагнозом СМА. Чаще патология встречается у мальчиков и мужчин.
Обнаруживается спинальная мышечная атрофия преимущественно у детей в раннем возрасте. Однако некоторые формы заболевания начинают проявляться только у подростков или уже взрослых людей. Коварство патологии заключается в том, что она постепенно, день за днем отбирает у больных то, что они сумели добиться.
Впервые патологию описал Г. Вердниг. Он обратил внимание на равностороннюю атрофию спинного мозга, его передних рогов, корешков периферических нервов в 1891 г. Уже в следующем году Дж. Хоффман сумел доказать, что речь идет о самостоятельном заболевании. В середине XX в. исследователи Е. Кугелберг и Л. Веландер описали патологию, которая возникает в позднем возрасте и имеет более благоприятный прогноз.
Дефицит белка SMN приводит к гибели моторных нейронов передних рогов спинного мозга. Вслед за нарушением иннервации начинается атрофия мышц. Spinal atrophy начинается с мускулатуры нижних конечностей. Мышечная атрофия затрагивает мускулы голени, бедра и стопы. Быстрее всего поражаются проксимальные группы мышц.
Тяжелее всего атрофия мышц проходит у детей раннего возраста. В процесс болезни (spinal atrophy) быстро включается мускулатура, ответственная за сосание, дыхание, глотание. Мышечная атрофия приводит к утрате основных жизненных навыков. При типе 1 СМА продолжительность жизни не превышает 1 года.
Поздние формы сма (spinal atrophy) протекают легче. Атрофия мышц начинается на нижних конечностях. Пациент жалуется на судороги и выраженную слабость. Может появляться тремор и фасцикуляции. Симптомы симметричны с обеих сторон. Постепенно атрофируются мышцы в области туловища. Респираторная и дыхательная мускулатура поражается редко.
Атрофия мышц верхних конечностей (при spinal atrophy) начинается последней. Мышечная атрофия сначала затрагивает плечи и надплечья. Позже недуг переходит на предплечья и мускулы кисти. На верхних конечностях также появляется тремор, болезненные спазмы.
Атрофия мышц у пациентов приводит к инвалидности. У каждого человека болезнь развивается по-разному. Мышечная атрофия может долго не проявлять себя при активных занятиях гимнастикой и лфк. Прогрессирующая атрофия мышечной ткани появляется только при 1 и 2 типе болезни.
В зависимости от сроков возникновения атрофии, также от течения, характера и степени тяжести спинальной мышечной атрофии у детей можно выделить следующую классификацию спинальных мышечных атрофий у детей: I тип – острая форма Верднига-Гоффмана; II тип – промежуточный (хронический инфантильный);
III тип – болезнь Кугельберга-Веландера (хроническая, ювенильная форма).
Клиническая картина

Характерно вялое, позднее шевеление плода во время беременности. После рождения ребенка отмечается «синдром гуттаперчевого ребенка», характеризующийся генерализованной мышечной гипотонией.
Уже в первые месяцы жизни появляются атрофии, а также фасцикулярные подергивания мышц туловища, спины, верхних и нижних конечностей (чаще в проксимальных отделах).
Важно
Наблюдается слабость межреберных мышц, вследствие чего грудная клетка выглядит уплощенной, мышц лица. В первые месяцы жизни у ребенка могут быть частые аспирации, дыхательные инфекции и пневмонии. Примерно в одном и том же возрасте (около шести месяцев) у ребенка заметна задержка двигательного развития.
Диагностика
Первый тип заболевания характеризуется быстро прогрессирующим злокачественным течением. Летальный исход обычно в полуторагодовалом возрасте. При биохимическом анализе крови отмечается незначительное повышение активности креатинфосфокиназы, альдолазы.
При электромиографическом исследовании виден «ритм частокола» – признак поражения передних рогов спинного мозга.

Дифференциальную диагностику первого типа спинальных мышечных атрофий следует проводить с врожденными миопатиями, со структурными миопатиями (болезнь центрального стержня, миотубулярная миопатия, пемалиновая миопатия), органическими ацидуриями.
Что повреждается в результате мутации
Из-за дефектного гена в организме нарушается выработка белка SMN — протеина выживаемости мотонейронов. Без этого белка мотонейроны – нервные клетки спинного мозга, отвечающие за координацию движений и мышечный тонус – отмирают, сигнал в мышцы ног, спины и отчасти рук не идёт.
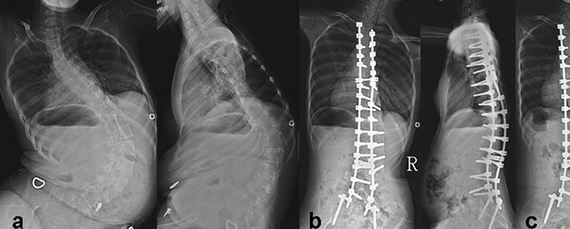
Без необходимого тонуса мышцы постепенно атрофируются. Отсутствие мышц пресса и спины приводит, кроме прочего, к обширным искривлениям позвоночника, а они – к проблемам с дыханием, которые из-за слабых мышц и так есть.
Болезнь может проявляться с первых месяцев жизни или в более позднем возрасте.
Изображение с сайта bfm.my
Симптоматика заболевания
Каждый вид СМА имеет свои особенные признаки, однако существуют некоторые симптомы, которые позволяют объединить разнородные заболевания в одну группу. Это:
- Нарастающая слабость мышц и их атрофия.
- При заболевании, проявившемся после 1-2 лет, заметна деградация уже достигнутых способностей, например, бега, ходьбы.
- Тремор пальцев. Дрожь наблюдается и на языке.
- Деформация скелета.
- Сохранность интеллектуального и психического здоровья у большинства больных.
Самая тяжелая клиническая картина развивается при sma 1 типа (спинально-мышечной атрофии у детей). Развивается СМА у ребенка с 6 месяцев. Характерны следующие признаки:
- Отсутствие активной моторики у младенцев со спинальной мышечной атрофией. Новорожденный вял, апатичен, плохо ест;
- Малыш не может перевернуться, сидеть, ползать;
- Снижается сосательный рефлекс, появляется частое поперхивание пищей, есть трудности с выделением секрета легких.
Прогноз при спинально мышечной атрофии у детей при таком типе болезни неблагоприятный. Диагностировать заболевание можно по характерным клиническим признакам, результатам генетического анализа и электромиографии.
Синдром Дубовица развивается в 6-18 месяцев. Здоровый ребенок, который мог ползать, сидеть, ходить, постепенно утрачивает свои навыки. Со временем появляется слабость дыхательной мускулатуры, деформируются конечности, позвоночник и грудная клетка.
Синдром Кугельберга-Веландера проявляется после 18 месяцев. Ребенок уже умеет стоять, ходить, выполняет сложные моторные действия. Больные не справляются с бегом, ходьбой по лестнице. Позже могут присоединяться нарушения жевания и глотания.
У взрослых мышечная атрофия появляется после 35 лет. Ее симптомы часто напоминают другие неврологические заболевания. Болезнь существенно затрудняет активность и приводит к инвалидности. Взрослый не может передвигаться без коляски, снижается его социализация. Болезнь практически не влияет на продолжительность жизни.
Генетический анализ позволяет подтвердить или опровергнуть наличие заболевания. Но у 5% пациентов с СМА он оказывается отрицательным из-за нетипичного расположения мутированного гена. В этом случае диагноз сложно подтвердить, основываясь только на клинической картине.
Болезни, с которыми следует дифференцировать СМА:
- Ботулизм детского возраста;
- Мышечная дистрофия Дюшенна;
- Нейропатия;
- Миопатия (метаболическая, врожденная);
- X-сцепленная СМА с расстройством дыхания.
Для дифференциальной диагностики используют электромиографию, компьютерную и магнитно-резонансную томографию, анализы крови на гормоны.
Выделяют 4 вида типичных СМА и несколько нетипичных форм болезни. Для генетического заболевания характерны мутации в 5 хромосоме в виде делеции гена SMN1. При нетипичных амитрофиях генотип может быть самым разнообразным.
Типичная СМА начинается в детском возрасте. А атипичная амиотрофия Кеннеди появляется в возрасте 30-50 лет. СМА болеют и мальчики, и девочки. А болезнь Кеннеди характерна только для мужского пола.
Дебют симптоматики появляется в возрасте после 2 лет. Ребенок жалуется на усталость при ходьбе, беге. Появляется шаткость и неустойчивость движений. Постепенно активность пациента снижается, ему недоступны сложные моторные навыки (бег, подъем по лестнице, спортивные игры).
Со временем пациент вынужден передвигаться в инвалидной коляске. У него развиваются контрактуры крупных суставов, атрофия мышц бедра, деформация грудной клетки и позвоночника. Это может сопровождаться появлением болевого синдрома. Корректируется своевременным назначением гимнастики и лфк.
Типичные признаки:
- Крыловидные лопатки;
- “Утиная” походка;
- Тремор языка и верхних конечностей;
- Отсутствие сухожильных рефлексов;
- Атрофия крупных групп мышц.
Амиотрофия Кеннеди
Болезнь характерна для взрослых лиц после 30 лет. Болеют в основном мужчины. У женщин случаев патологии не описано. Первые проявления характеризуются усталостью икроножных мышц и мускулов бедра. Пациент замечает, что не может долго ходить и стоять.
Атрофия прогрессирует медленно. Около 10 лет после появления первых признаков патологии больные могут продолжать привычный образ жизни. Позже заболевание переходит на верхние конечности: появляется тремор, атрофируются мускулы головы и шеи.
Для амиотрофии Кеннеди характерны эндокринологические изменения. У взрослых мужчин определяется недостаток половых гормонов, происходит атрофия яичек и снижение полового влечения. Поражается ткань поджелудочной железы, развивается сахарный диабет.
Дистальная СМА развивается в возрасте 20-50 лет. При заболевании происходит атрофия мышц дистальных отделов верхних конечностей (кистей, предплечий). Позже развиваются нарушения на ногах. В них вовлекаются стопы и голени. Со временем идет атрофия всей мускулатуры конечностей.
СМА Вюльпиана характеризуется атрофией мышечных групп лопаток и мышц голеней. Синдром развивается после 20 лет, но вероятность атрофии сохраняется до 40 лет. При болезни появляется ограничение подвижности плечевых суставов. Лопатки выступают и напоминают крылья. Отсюда характерный признак СМА Вюльпиана – симптом «крыловидных» лопаток.
СМА этого типа позволяет 30-40 лет оставаться активным и сохранять подвижность больному человеку. Только при нарушении функции разгибателей стоп и атрофии мышц голеней теряется его способность к передвижению. В целом, это благоприятный тип течения амиотрофии, который не ведет ранней инвалидности и не становится причиной смерти.
СМА 1 и СМА 2 имеет разные симптомы и признаки.
Какие существуют отдаленные последствия черепно мозговой травмы и как максимально себя обезопасить от получения повреждения головы.
Разрыв вен и сосудов головного мозга провоцирует такое заболевание, как субдуральная гематома головного мозга. В чем сложность лечения и диагностики заболевания.
Первые симптомы выявляют еще во время беременности по слабому шевелению плода.
От чего зависит степень тяжести болезни
За выработку белка SMN отвечают два гена — SMN1 и SMN2.
При этом SMN1 – основной «заказчик» данного белка, а SMN2 – дополнительный, он вырабатывает белок в количестве, недостаточном для нормальной работы организма. В случаях, когда в геноме человека SNM1 отсутствует, SNM2 начинает выполнять замещающие функции, но никогда не может полностью восполнить недостачу.
Копий SMN2 в геноме бывает до восьми. От имеющегося у человека числа копий SMN2 и зависит тяжесть состояния больного. Такой сложный механизм болезни приводит к тому, что СМА имеет несколько форм, и состояние больных – очень разное.
Причины и механизм развития заболевания
Спинальная амиотрофия развивается из-за мутировавшего SMN гена пятой хромосомы. Если оба родителя – его носители, существует 25%-ная вероятность, что ребенок родится больным.
Мутация гена SMN приводит к нарушению синтеза белка, в результате чего происходит разрушение мотонейронов спинного мозга. Нервные импульсы не проходят к мышцам, которые из-за бездействия атрофируются, человек теряет способность двигаться.
Считается, что теряет работоспособность сначала глубоко расположенная мускульная ткань.
Какие формы СМА существуют?
Существует 4 типа СМА, различающиеся степенью тяжести и возрастом, в котором впервые проявляется заболевание.
СМА I, болезнь Верднига-Гоффмана. Самая тяжёлая форма болезни, проявляется у младенцев от 0 до 6 месяцев. Дети с этой формой с рождения имеют трудности с дыханием, сосанием и глотанием, а также не осваивают самые простые контролируемые движения — не держат голову, не сидят самостоятельно. Ранее считалось, что большинство (80%) не доживают до двух лет. Сейчас благодаря новым стратегиям ИВЛ и зондовому кормлению срок жизни можно продлить ещё на несколько месяцев.
СМА II, болезнь Дубовица. Первые проявления болезни в 7-18 месяцев. Человек с таким типом СМА может есть и сидеть, но не ходит самостоятельно. Продолжительность жизни зависит от степени поражения мышц, обеспечивающих дыхание.
СМА III, болезнь Кюгельберга-Веландер. Болезнь впервые проявляется после полутора лет. Такие больные могут стоять (испытывая боль), но не ходят. На продолжительность жизни СМА III типа, как правило, не влияет, но сильно ухудшает её качество.
СМА IV, этот тип называется ещё «взрослой СМА», поскольку болезнь проявляется обычно в возрасте после 35 лет.Симптомы – мышечная слабость, сколиоз и тремор. Кроме того, развиваются контрактуры суставов (ограничения подвижности в суставах) и нарушения метаболизма.Прогрессирование заболевания не очень быстрое, сначала мышечная слабость затрагивает мышцы ног, затем – рук.

СМА, связанные с нарушением гена SMN, в медицинской литературе называют проксимальными — они составляют 95% от всех спинальных амиотрофий. СМА, не связанных с геном SMN, довольно много, но встречаются они редко. К ним относится, например, болезнь Кеннеди. Исследования 1990-х годов показали, что болезнь Кеннеди не связана не с поломкой гена SMN1, но с другими генетическими мутациями, приводящими к нарушению усвоения белка SMN. Болезнь проявляется у людей старше 35 лет. Для СМБА характерна, в основном, слабость конечностей.
Один из видов СМА, не связанный с геном SMN, называется болезнь Кеннеди. То, что эту болезнь до сих пор иногда относятся к СМА – анахронизм. В конце 1960-х, когда было выполнено подробное описание этой атрофии, её посчитали разновидностью СМА, так как при ней поражаются те же нервы и мышцы, что и при трёх типах СМА (но в гораздо меньшей степени).
Выделяю такие виды:
- ранняя детская или СМА 1 – признаки заболевания проявляются до 6 месячного возраста;
- поздняя форма или СМА 2 – симптомы появляются после 6 месяцев до 1 года.
Диагностика
Наиболее точным методом определения спинально-мышечной атрофии у детей является анализ ДНК. Он проводится как у родившегося малыша, так и во время внутриутробного развития. Дополнительно проводятся следующие исследования:
- Анализ на биохимию. Целью является выяснение уровня ферментов: ананинаминотрансферазы, лактатдегидрогеназы, креатинкиназы. Нормальное их содержание позволяет исключить подозрения на прогрессирующую дистрофию мышц.
- Электрофизиологическое исследование. Метод направлен на регистрацию биоэлектрической активности. Патологию характеризует ритм «частокола».
- МРТ. Назначается для обнаружения признаков атрофии мышц.
- Микроскопия спинного мозга. Отмечаются признаки дегенеративных процессов в клетках нервных отростков. Они сморщиваются, разбухают, при этом глиальные волокна имеют плотную структуру.
- Тандемная масс-спектрометрия. Исследование помогает уточнить уровень аминокислот и белка СМН.
- Гистологическое исследование поперечнополосатых мышц. По результатам будут видны группы мелких волокон.
Если у молодых людей, планирующих рождение ребенка, есть родственники с патологией СМА, им рекомендовано пройти генетическую экспертизу.
При спинальной амиотрофии Вердника диагностика заключается в проведении генетического анализа, выявляя мутации или делецию гена SMN.
При обнаружении делеции теломерной копии SMNt диагноз считают подтвержденным.
В случае отсутствия делеции проводят дополнительные исследования:
- электонейромиографию;
- исследование нервной проводимости;
- тест на креатинкиназу;
- биопсию мышц и нервной ткани.
При нормальных показателях фермента креатинкиназы проводят подсчет копий SMNc. В случае единственной копии идентифицируют точечную мутацию, принимая окончательное решение.
Как это лечат?
Изображение с сайта asesoramientopsicologicoalicante.es
На сегодняшний момент радикального лечения от СМА не существует.
Международной корпорацией «Биоген» был разработан препарат «Спинраза», который значительно улучшил состояние больных, к которым применялся во время тестирования. В настоящее время препарат одобрен к применению в США, в Европе ориентировочная стоимость годового курса, по подсчётам компании, будет составлять около 270 тысяч евро, в России препарат не сертифицирован. Лечение пожизненное.
У моего племянника диагноз “Спинальная амиотрофия Вердинга-Гофмана” с рождения, сейчас ему 4 года. Сноха нашла клинику в Китае, где берутся за лечение. У нас возникли сомнения, стоит ли им верить, может это мошенники. Посоветуйте, пожалуйста, как нам быть. Прочитайте нашу переписку, и скажите, пожалуйста, Ваше мнение об этом.
Здравствуйте, Оксана!
Меня зовут Николай, я сотрудник международного отдела медицинского центра Wu stem cells, Пекин, Китай,и курирую русскоязычных пациентов.

У наших специалистов богатый опыт лечения детей с этим заболеванием. Мы будем рады вам помочь!
Совет
Спасибо за высланную информацию о вашем сыне, в течении 1-3 рабочих дней наш главный врач рассмотрит её,
и я вышлю вам его ответ, в котором, в случае положительного решения, будет подробно указана
программа лечения, длительность курса, ожидаемые результаты, и точная стоимость.
Прогноз
То, как будет развиваться болезнь, сколько лет проживет ребенок, зависит от ее типа.
При атрофии типа один прогноз крайне неблагоприятен. Около 50% малышей не доживают и до двух лет. Не больше 10% детей с болезнью Верднига-Гоффмана могут дожить до пяти лет. Причиной гибели чаще всего становится воспаление легких, остановка дыхания, сердца.
Пациенты, которым диагностирована болезнь Дубовица, живут в среднем до 10, иногда 12 лет. Около 30% малышей умирают, не достигнув четырех лет.

При SMA III типа детская смертность встречается реже. У многих пациентов симптомы появляются в предподростковом-подростковом возрасте. Через несколько лет они перестают ходить. Далее, по нарастающей, отмечается атрофия мышц внутренних органов, в том числе дыхательных.
Считается, что заболевание IV типа не влияет на продолжительность жизни, тем не менее, оно ведет к инвалидизации.
Можно ли помочь больным СМА и как именно?
Вылечить болезнь пока нельзя, но можно облегчать состояние больных СМА, то есть различными способами компенсировать проявления болезни.
При тяжёлых типах СМА больным приходится помогать дышать и глотать. Поэтому им жизненно необходимы мобильные аппараты ИВЛ, аспираторы-откашливатели, мешки Амбу.
Ещё детям со СМА очень нужна помощь волонтёров, способных хоть на короткое время подменить родителей.
Детям, больным СМА, помощь может понадобиться в любой момент, поэтому мамы и папы всегда начеку и сами осваивают навыки реанимации, необходимые на случай, если ребенок внезапно перестал дышать.

Менее тяжёлым больным нужны лекарства, облегчающие дыхание, корсеты, коляски и другие приспособления, облегчающие перемещение и жизнь людей со слабыми мышцами.
Болезнь, продолжающаяся много лет, выматывает, поэтому пациентам, особенно взрослым, часто необходима помощь психолога.
Несмотря на то, что впервые СМА была описана австрийским неврологом Гвидо Верднигом и немецким неврологом Джоханном Хоффманном ещё в начале 1890-х годов, полностью понять природу заболевания удалось только в конце XX века. Ген SMN1 был открыт в 1995 году. Чтоб подтвердить диагноз СМА, нужен генетический тест.
В России соответствующие генетические тесты стали доступны в начале 2000-х годов. Генетический тест на СМА возможно сделать по ОМС, однако на практике не слишком много врачей знают этот редкий диагноз и направляют больных на соответствующее исследование. Стоимость такого тестирования в коммерческих лабораториях Москвы – порядка 6 тысяч рублей.
Отсутствие специальной диагностики привело также к путанице в диагнозах. Большинство больных СМА в России не выявлены, у многих выявленных в качестве диагноза записана «болезнь Верднига-Гоффмана», хотя не у всех из них (особенно взрослых) в действительности именно этот тип болезни.
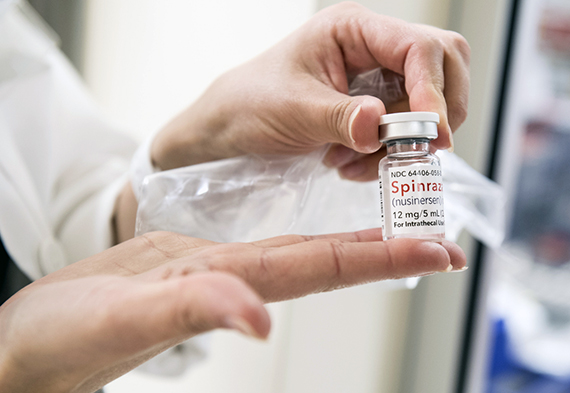
Препарат «Спинраза», значительно улучшающий состояние больных. Фото с сайта healthbeat.spectrumhealth.org
С учётом частоты заболевания, количество больных СМА в России должно составлять от семи до двадцати четырёх тысяч человек. На сегодняшний день в реестре пациентов фонда «Семьи СМА» находится около 400 человек.
Благотворительный фонд «Вера», детский хоспис «Дом с маяком», благотворительный фонд «Детский паллиатив», благотворительный фонд «Семьи СМА», детская паллиативная служба «Милосердие».
С 2014 года в Москве развивается совместный проект службы «Милосердие» и фонда «Семьи СМА» «Клиники СМА» На встречах, которые проходят раз в месяц, больные могут получить консультации пульмонолога, ортопеда, физиотерапевта и психолога. В последнее время часть встреч ориентированы и на нужды взрослых пациентов.
Профилактика
Мер, направленных на профилактику и предотвращение развития СМА, не существует. Женщина, ожидающая рождения ребенка, может заподозрить проблему, обратив внимание на слабость шевелений плода. Проведенный ДНК-анализ может подтвердить или развеять подозрения. При необходимости проводится медицинская комиссия, которая может порекомендовать прерывание беременности. Врач обязательно рассказывает о заболевании, его течении и последствиях.
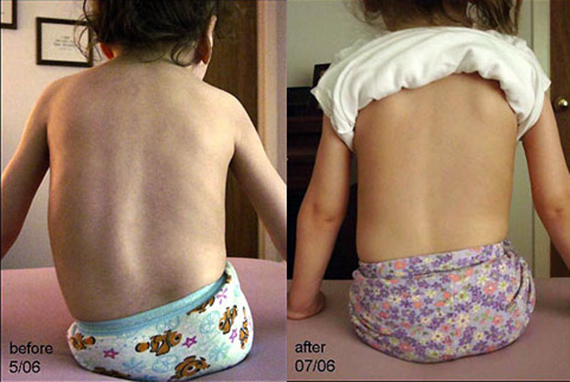
После диагностики заболевания у уже родившегося ребенка его окружают заботой и вниманием. Использование системы искусственной вентиляции легких, отсасывателей мокроты, специальных приспособлений для движения малыша, который может передвигаться, помогают улучшить качество жизни и помочь ребенку жить.
Спинальная амиотрофия – опасная, пока не поддающаяся лечению патология. Она характеризуется атрофией мышц. Возникает в разном возрасте. Прогноз в большинстве случаев неблагоприятный.
Для подготовки статьи использовались следующие источники:Селиверстов Ю. А., Клюшников С. А., Иллариошкин С. Н. Спинальные мышечные атрофии: понятие, дифференциальная диагностика, перспективы лечения // Журнал Нервные болезни — 2015Лепесова М. М., Ушакова Т. С., Мырзалиева Б. Д. Дифференциальная диагностика спинальной мышечной амиотрофии первого типа // Вестник Алматинского государственного института усовершенствования врачей — 2016
Можно ли заболеть СМА из-за прививок?
В Европе и США связь между прививками и проявлением болезни не прослежена.
Понять, есть ли связь между СМА и прививками, может объяснение разницы между СМА и полиомиелитом. Полиомиелит – инфекционное заболевание, когда от инфекции повреждается организм изначально здорового ребёнка. Ребёнок со СМА, родившийся с повреждённым геномом, внешне может выглядеть здоровым, но на самом деле он уже болен, просто симптомы его болезни проявляются постепенно.
Большинство проявлений СМА связаны с освоением первых двигательных навыков. Первые проявления болезни совпадают по времени с несколькими возрастными прививками. В итоге человек и его родные могут утверждать, что он «заболел от прививки», но на самом деле у него просто проявились признаки болезни, которая уже была.
Полезные ресурсы про СМА
Сайт фонда
«Семьи СМА»
Фонд публикует информацию о болезни и свежие новости о лечении СМА в России. Много новостей выходит на странице фонда в ФБ.
Американская ассоциация мышечных дистрофий (англоязычный),
Форум о миопатиях (российский).
Фонд «Дети со СМА» (Украина) (на сайте есть форум).
Благодарим за предоставленную информацию фонд «Семьи СМА» и лично Ирину Старову-Кислину.





